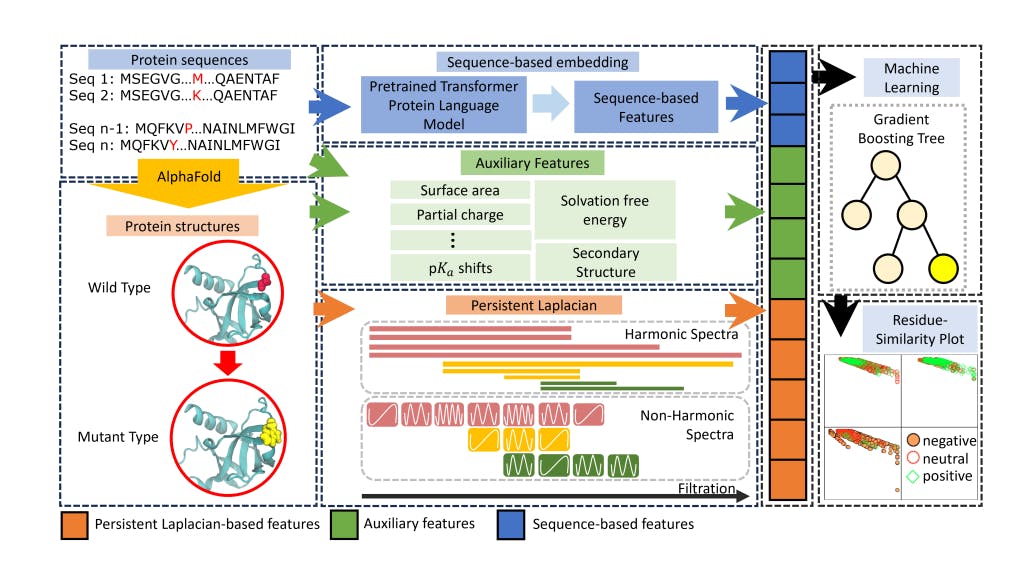
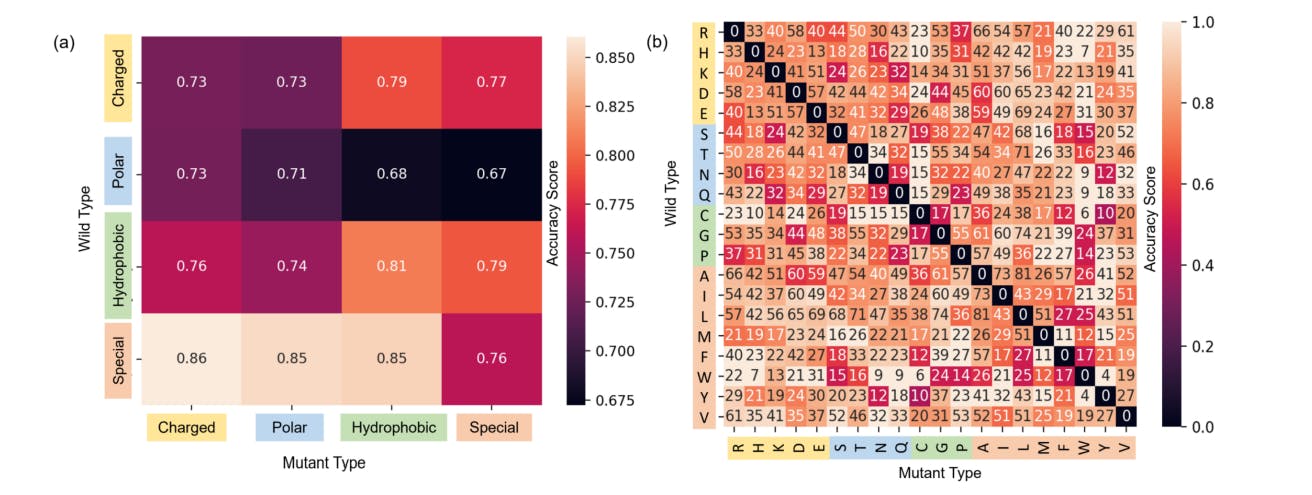
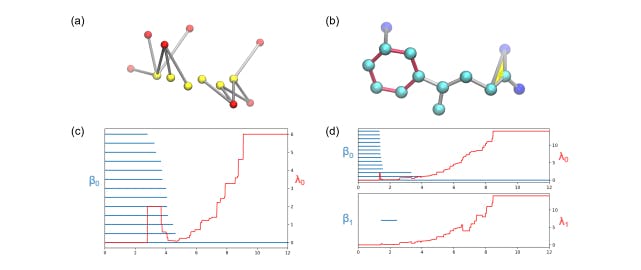
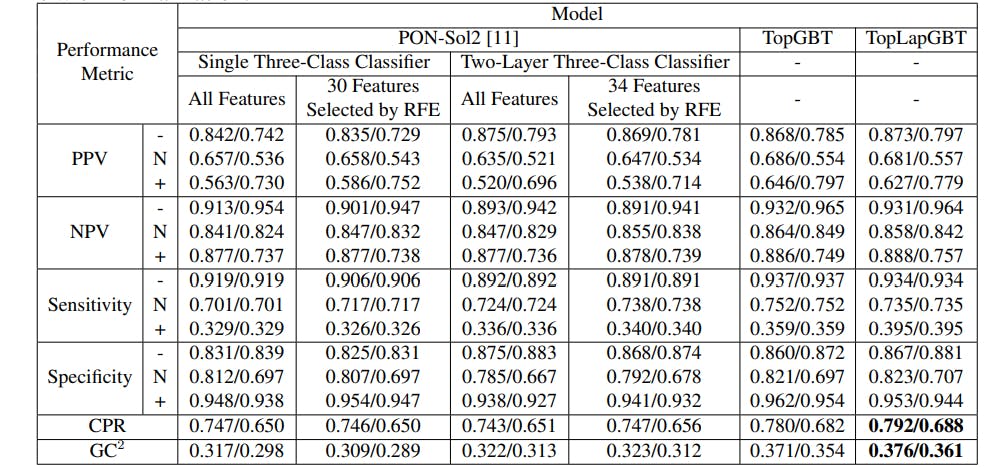
167 reads
TopLapGBT: A Unified Approach for Enhanced Protein Solubility Prediction
by byMutation Technology Publications@mutation
byMutation Technology Publications@mutation
Mutation: process of changing in form or nature. We publish the best academic journals & first hand accounts of Mutation
February 16th, 2024





Audio Presented by
Mutation: process of changing in form or nature. We publish the best academic journals & first hand accounts of Mutation


Mutation: process of changing in form or nature. We publish the best academic journals & first hand accounts of Mutation
About Author

Mutation: process of changing in form or nature. We publish the best academic journals & first hand accounts of Mutation
Comments